' fill='none' stroke='%23304560' stroke-miterlimit='10' stroke-width='2'/%3e%3c/svg%3e)

ASRtriala: leveraging ASReml-R for powerful trial analysis
Dr. Giovanni Galli
23 March 2022
Performing statistical analysis in plant breeding is usually an overwhelming task. For analysis of field trials there are many difficult questions to answer before we even start, such as:
- Are my data reliable?
- Should I analyze the trials individually?
- Which model should I use?
- What terms do I use for spatial analyses?
- How do I know I have “the best possible” model given the realm of choices?
- Which metrics should I use to compare models?
- How do I combine the trials into a multi-environment model?
- What is the best way to consider genotype by environment interaction?
- Is there anything else I should do after my model is fitted?
And this list goes on and on.
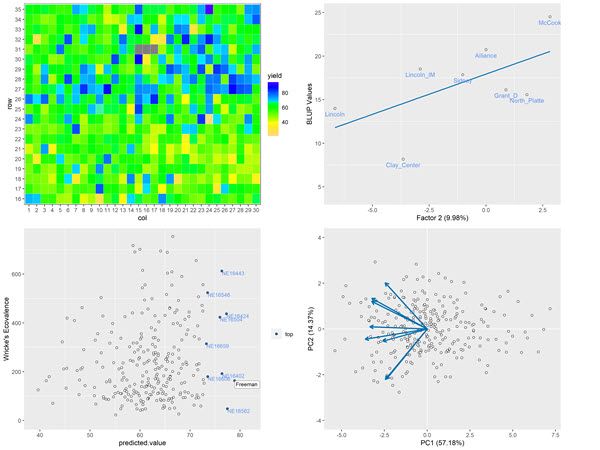
We developed ASRtriala (ASR trial analysis) with the above questions in mind. ASRtriala is the newest member of VSNi’s companion packages for ASReml-R. This is a free to use R library which can be downloaded from this web page. The package is aimed at plant breeders with the purpose of improving their experience, providing robust and in-depth analyses of their single and multi-environment trials. The main capabilities of the package include:
- Auditing/preparing single-trial data and multi-environment trial data.
- Fitting/selecting a single-trial model (spatial and non-spatial) using ASReml-R.
- Fitting/selecting a multi-environment trial model using ASReml-R.
- Enhancing output from multi-environment trial models.
The library provides building blocks (functions) that can be put together in a flexible way for a complete data-to-decision workflow for spatial and non-spatial single and multi-environment trial analysis. It allows for a simpler, easier, and robust flow from raw data to genotype selection with a seamless and efficient two-stage (single trial followed by multi-environmental trial) analysis.
We invite you to try this free library and check out the user guide for a more detailed walkthough and technical explanations.
About the author
Dr. Giovanni Galli is an Agronomist with an M.Sc. and Ph.D. in Genetics and Plant Breeding from the University of São Paulo (USP)/Luiz de Queiroz College of Agriculture (ESALQ). He currently works as a Statistical Consultant at VSN International, United Kingdom. Dr. Galli has experience in field trials, quantitative genetics, conventional and molecular breeding (genomic prediction and GWAS), machine learning, and high-throughput phenotyping.
Popular



Related Reads